Quels sont les signes ?
Les symptômes de la cystinose varient en gravité, mais ils incluent généralement un retard de croissance, des problèmes rénaux, une sensibilité à la lumière (photophobie), des dommages aux yeux tels que la formation de cristaux dans la cornée (kératopathie cornéenne), des troubles endocriniens, et une dégradation progressive de la fonction rénale. Si elle n’est pas traitée, la cystinose peut donc entraîner une insuffisance rénale et d’autres problèmes de santé graves. Notons que ces symptômes sont en grande partie pris en charge aujourd’hui.
La Cystinose nephropatique – Forme infantile
Symptômes rénaux
Lorsqu’elle n’est pas traitée, la cystinose néphropathique est marqué par une atteinte rénale précoce – dans la première année – et sévère, le syndrome de Fanconi.
Les symptômes cliniques associent une polyurie (besoin d’uriner trés fréquent), une soif intense, des vomissements et éventuellement des épisodes de déshydratation.
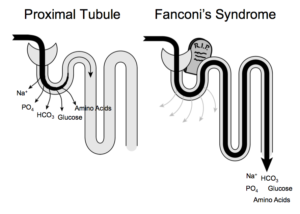 Ce syndrome de Fanconi est caractérisé par une déficience au niveau des tubules proximaux du rein qui sont les éléments où a lieu la filtration du sang. Ils présentent une insuffisance généralisée à réabsorber l’eau, les électrolytes, le bicarbonate, le glucose, le phosphate, les protéines, etc. Cela provoque principalement une hyperaminoacidurie (présence d’acides aminés dans les urines) et une glycosurie (présence de sucres dans les urines). Le bicarbonate est quant à lui considérablement moins réabsorbé d’où une chute de sa concentration dans le sérum. Cela crée une acidose métabolique qui est partiellement responsable de la faible croissance des enfants malades.
Ce syndrome de Fanconi est caractérisé par une déficience au niveau des tubules proximaux du rein qui sont les éléments où a lieu la filtration du sang. Ils présentent une insuffisance généralisée à réabsorber l’eau, les électrolytes, le bicarbonate, le glucose, le phosphate, les protéines, etc. Cela provoque principalement une hyperaminoacidurie (présence d’acides aminés dans les urines) et une glycosurie (présence de sucres dans les urines). Le bicarbonate est quant à lui considérablement moins réabsorbé d’où une chute de sa concentration dans le sérum. Cela crée une acidose métabolique qui est partiellement responsable de la faible croissance des enfants malades.
Lorsqu’il atteint le tubule distal, cet excès de bicarbonate améliore l’excrétion de potassium entraînant une hypokaliémie sévère (faibles niveaux de potassium dans le sérum) et donc un risque de dysfonctionnement cardiaque. Des protéines de faible et de haut poids moléculaire sont également fréquemment trouvées dans les urines des patients. Ce dommage tubulaire rénal est en général irréversible.
En plus du syndrome de Fanconi, il y a une perte progressive de la fonction glomérulaire (filtration par le rein). Celle-ci est normale puis elle se détériore progressivement provoquant une insuffisance rénale avant l’adolescence. Cependant, l’évolution de la maladie rénale diffère selon les patients. La fonction rénale va se stabiliser pour certains malades, tandis qu’elle va se détériorer pour d’autres.
Sans traitement, l’évolution se fait inéluctablement vers l’apparition d’une insuffisance rénale chronique, aboutissant à l’insuffisance rénale terminale aux alentours de dix ans.
Symptômes extra-rénaux
De nombreuses manifestations extra-rénales surviennent au cours de l’évolution de la maladie, dues à l’accumulation de cystine dans les différents organes.
Le retard de croissance est majeur sans traitement, la taille définitive des adultes étant en moyenne de 136,5 cm chez les hommes et 124 cm chez les filles.
L’atteinte oculaire est précoce, marquée par une photophobie douloureuse (sensation douloureuse des yeux provoquée par la lumière) et un larmoiement dus à la présence de cristaux de cystine dans la cornée et dans la conjonctive, cristaux aisément détectables par l’examen à la lampe à fente dès l’âge d’un an. L’atteinte rétinienne (zones de dépigmentation irrégulières) est également précoce, et peut aboutir à une baisse de la vision, généralement à partir de l’âge de dix ans. Sans traitement, l’atteinte oculaire progresse vers une perte de la vision.
Une hépatomégalie (augmentation de la taille du foie), due à l’infiltration des cellules de Kupffer par les cristaux de cystine, est retrouvée dans 40% des cas et peut aboutir au développement d’une hypertension portale.
L’hypothyroïdie biologique est également commune, même si les symptômes cliniques d’hypothyroïdie sont rarement présents. Elle contribue néanmoins à l’altération de la croissance.
Un hypogonadisme (défaut de l’appareil reproducteur- fertilité) est fréquent chez le garçon, lié à l’accumulation de cystine dans les testicules.
Un diabète insulino-dépendant, généralement transitoire survenant après transplantation rénale sous doses élevées de corticoïdes, et une insuffisance pancréatique externe peuvent survenir du fait de la surcharge pancréatique.
L’atteinte musculaire est fréquente. Il s’agit initialement d’une faiblesse musculaire liée à l’hypokaliémie ( baisse de la concentration de potassium dans le sang qui peut avoir des conséquences graves sur le fonctionnement musculaire et cardiaque) et à un déficit en carnitine (peut entrainer une faiblesse musculaire ou faible tonus musculaire, fatigue, irritabilité, retard de motricité, hypoglycémie (si le foie est affecté), essoufflement (si le cœur est affecté)). Les enfants plus âgés peuvent développer une véritable myopathie liée à la surcharge en cystine des cellules musculaires.
Sans traitement, l’atteinte du système nerveux central est une complication tardive survenant après l’âge de 20 ans. Elle associe des difficultés à la marche, des troubles de la déglutition et de la parole et une détérioration des fonctions cognitives, mettant à terme en jeu le pronostic vital.
La Cystinose – Formes juvéniles et adultes
La forme juvénile débute plus tardivement, l’âge de début de la maladie pouvant être très étalé, des débuts de 2 à 26 ans étant rapportés dans la littérature. Les symptômes tubulaires sont très atténués voire absents et la maladie peut se manifester uniquement par une atteinte glomérulaire évoluant très progressivement vers l’insuffisance rénale terminale. Les manifestations extra-rénales sont elles aussi moins sévères et d’apparition beaucoup plus tardives, voire même absentes, en dehors de l’atteinte oculaire. Quant à la forme adulte, elle se traduit par une atteinte oculaire isolée, parfois uniquement découverte par l’examen à la lampe à fente.
Les problèmes neurologiques se produisent chez 5 à 10 % des adultes malades. Ils sont hétérogènes et incluent une augmentation du volume du liquide céphalo-rachidien, des troubles cognitifs(1) des troubles de la personnalité… Ces troubles neurologiques sont faiblement documentées, une étude sur l’observance et la compréhension des mécanismes physiopathologiques liés aux complications neurologiques de la cystinose est mené en France (projet CrYStobs).















")
