


La cystinose est une maladie autosomique récessive qui est due à une accumulation de cystine dans les lysosomes. Cela concerne les cellules de tous les tissus et organes avec une plus grande sensibilité de la part du tissu rénal. C’est en fait l’absence de cystinosine, un transporteur de la cystine situé dans la membrane du lysosome, qui est responsable de cette accumulation, la cystine n’étant plus transportée hors du lysosome.
Il existe trois formes de cystinose :
* La cystinose infantile classique ou néphropathique : forme la plus sévère qui, sans traitement, conduit à une insuffisance rénale dans la première décennie de la vie.
* La forme intermédiaire de la cystinose. Elle présente les mêmes manifestations que la forme néphropathique mais celles-ci apparaissent plus tardivement, généralement autour de l’adolescence.
* La cystinose oculaire ou non néphropathique. Elle est caractérisée par une atteinte oculaire uniquement. Les reins sont épargnés.
Trouvée dans tous les groupes ethniques, cette maladie a une prévalence comprise entre une naissance sur 200 000 et une naissance sur 100 000 selon les pays (En France, l’estimation est de 5 nouveaux cas par an). 95 % des personnes atteintes de cystinose ont la forme néphropathique (infantile).
LA CYSTINOSE NEPHROPATHIQUE
Lorsqu’elle n’est pas traitée, la cystinose néphropathique est associée à une faible croissance et au syndrome de Fanconi (voir schéma ci-contre) dès les premières années de vie.
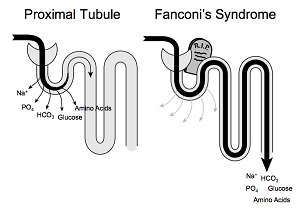 Ce syndrome de Fanconi est caractérisé par une déficience au niveau des tubules proximaux du rein qui sont les éléments où a lieu la filtration du sang. Ils présentent une insuffisance généralisée à réabsorber l’eau, les électrolytes, le bicarbonate, le glucose, le phosphate, les protéines, etc. Cela provoque principalement une hyperaminoacidurie (présence d’acides aminés dans les urines) et une glycosurie (présence de sucres dans les urines). Le bicarbonate est quant à lui considérablement moins réabsorbé d’où une chute de sa concentration dans le sérum. Cela crée une acidose métabolique qui est partiellement responsable de la faible croissance des enfants malades.
Ce syndrome de Fanconi est caractérisé par une déficience au niveau des tubules proximaux du rein qui sont les éléments où a lieu la filtration du sang. Ils présentent une insuffisance généralisée à réabsorber l’eau, les électrolytes, le bicarbonate, le glucose, le phosphate, les protéines, etc. Cela provoque principalement une hyperaminoacidurie (présence d’acides aminés dans les urines) et une glycosurie (présence de sucres dans les urines). Le bicarbonate est quant à lui considérablement moins réabsorbé d’où une chute de sa concentration dans le sérum. Cela crée une acidose métabolique qui est partiellement responsable de la faible croissance des enfants malades.
 Lorsqu’il atteint le tubule distal, cet excès de bicarbonate améliore l’excrétion de potassium entraînant une hypokaliémie sévère (faibles niveaux de potassium dans le sérum) et donc un risque de dysfonctionnement cardiaque. Des protéines de faible et de haut poids moléculaire sont également fréquemment trouvées dans les urines des patients. Ce dommage tubulaire rénal est en général irréversible.
Lorsqu’il atteint le tubule distal, cet excès de bicarbonate améliore l’excrétion de potassium entraînant une hypokaliémie sévère (faibles niveaux de potassium dans le sérum) et donc un risque de dysfonctionnement cardiaque. Des protéines de faible et de haut poids moléculaire sont également fréquemment trouvées dans les urines des patients. Ce dommage tubulaire rénal est en général irréversible.
En plus du syndrome de Fanconi, il y a une perte progressive de la fonction glomérulaire. Celle-ci est normale puis elle se détériore progressivement provoquant une insuffisance rénale avant l’adolescence. Cependant, l’évolution de la maladie rénale diffère selon les patients. La fonction rénale va se stabiliser pour certains malades, tandis qu’elle va se détériorer pour d’autres.
Durant l’enfance, une atteinte oculaire apparait progressivement. Celle-ci est due à un dépôt de cystine dans la cornée (sous forme de cristaux) qui associé à une atteinte de la rétine va entrainer une photosensibilité (sensation douloureuse des yeux provoquée par la lumière) et des conjonctivites. Sans traitement, l’atteinte oculaire progresse vers une perte de la vision.
LE DIAGNOSTIC
Le diagnostic biochimique repose sur le dosage de cystine intra-leucocytaire. Pour tester la présence ou l’absence de mutations, un diagnostic moléculaire par recherche de mutations est possible. Les familles avec un antécédent de cystinose peuvent par ailleurs avoir recours à un diagnostic prénatal qui est réalisé en mesurant le taux de cystine dans les cellules prélevées du liquide amniotique et mises en culture ou dans les cellules des villosités choriales (futur placenta).
Chez toutes les personnes atteintes de cystinose, des mesures appropriées doivent être prises (tests de la fonction rénale, étude des fonctions thyroïdiennes, évaluation ophtalmologique…).
LES TRAITEMENTS
Sans traitement, la cystine s’accumule dans pratiquement tous les organes et tissus et y forme des cristaux du fait de sa faible solubilité. Plusieurs complications peuvent accompagner la détérioration des tissus comme une hypothyroïdie, un petit retard de puberté, une hypertension intracrânienne bénigne… Dans la cystinose, la cognition(1) est normale bien que des anomalies neurocomportementales s’expriment chez certains malades.
Il existe plusieurs traitements pour soigner les individus atteints de cystinose. Le syndrome de Fanconi est traité en remplaçant les pertes rénales, en laissant un accès libre à l’eau et en donnant un complément en citrate pour alcaliniser le sang. Une hormone de croissance peut être prescrite aux enfants pour améliorer leur croissance et augmenter la réabsorption du phosphate. Il est toutefois important de noter que les personnes bien traitées grandissent généralement bien et ne nécessitent donc pas d’hormones de croissance. Pour soulager la protéinurie glomérulaire sévère, des inhibiteurs de l’enzyme de conversion de l’angiotensine peuvent être utilisés. La transplantation rénale est généralement indiquée quand la clairance de la créatinine est très faible et que l’azotémie et l’hypertension progresse rapidement. Une allogreffe rénale soigne le syndrome de Fanconi mais pas les autres complications multisystémiques.
Un traitement appauvrissant en cystine est actuellement donné oralement sous forme de cystéamine bitartrate. Cela a révolutionné la gestion et le pronostic de la cystinose néphropathique. La cystéamine bitartrate entre dans le lysosome chargé en cystine, réagit avec celle-ci et forme un composé. Ce composé peut ensuite sortir du lysosome. La cystéamine épuise les lysosomes de plus de 90 % de leur contenu en cystine. Il faut cependant qu’elle soit donnée tôt, régulièrement et à fortes doses pour retarder la progression de la maladie. Les effets secondaires sont sans gravité (nausée, troubles digestifs, goût désagréable…) cependant l’odeur soufrée induite par le traitement, imposante chez certains patients, peut entrainer des difficultés sociales importantes. A l’adolescence, cette gêne peut être à l’origine de difficultés d’observance du traitement de la part du malade. En addition du traitement oral de cystéamine, un traitement sous forme de collyre est également indiqué afin de prévenir/modérer les atteinte oculaires.
La cystéamine est désormais un traitement de choix pour la cystinose à travers le monde. Cependant, la majorité des patients qui n’ont pas reçu suffisamment tôt, de façon assidue et sur le long terme, de cystéamine souffriront de plusieurs complications majeures. Elles incluent différents systèmes organiques comme le système ophtalmologique, musculo-squelettique, gastro-intestinal, endocrinien, néphrologique, cardiovasculaire, neurologique… Les problèmes neurologiques se produisent chez 5 à 10 % des adultes malades. Ils sont hétérogènes et incluent une augmentation du volume du liquide céphalo-rachidien, des troubles cognitifs(1) des troubles de la personnalité… Ces troubles neurologiques sont faiblement documentées, une étude sur l’observance et la compréhension des mécanismes physiopathologiques liés aux complications neurologiques de la cystinose est actuellement en cours en France (projet CrYStobs).
Delphine GENEVAZ
(1) Ensemble des processus mentaux qui se rapportent à la fonction de connaissance






")






