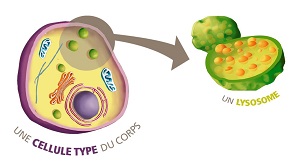
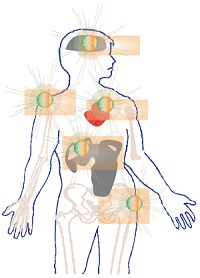
Sous l’appellation de « maladies lysosomales » sont regroupées une cinquantaine d’affections handicapantes de l’enfant et de l’adulte dont le point commun est une déficience génétique induisant un défaut de fonctionnement au niveau du lysosome.
- L’association
Une association de parents et patients adultes
Représenter les malades et familles
Soutenir la recherche
- Les maladies Lysosomales
- La recherche
L'espoir de guérison
Une expertise partagée

- Nous soutenir
J'agis avec vml - bénévolat
- Les Lyso-Gestes, des idées pour Agir !
- Je crée une page de collecte (challenge - anniversaire ...)
- Je veux être bénévole
- Je veux proposer un projet, une manifestation
- Je propose une Balade du Lysosome
- Je veux être Ambassadeur LysoSolidarité
- Je rejoins le TeamSport du Lysosome
- La Boutique du Lysosome
- Actualités
- Contact
- L’association
Une association de parents et patients adultes
Représenter les malades et familles
Soutenir la recherche
- Les maladies Lysosomales
- La recherche
L'espoir de guérison
Une expertise partagée
- Nous soutenir
J'agis avec vml - bénévolat
- Les Lyso-Gestes, des idées pour Agir !
- Je crée une page de collecte (challenge - anniversaire ...)
- Je veux être bénévole
- Je veux proposer un projet, une manifestation
- Je propose une Balade du Lysosome
- Je veux être Ambassadeur LysoSolidarité
- Je rejoins le TeamSport du Lysosome
- La Boutique du Lysosome
- Actualités
- Contact