Qu’est ce que la maladie ?
C’est une maladie héréditaire à transmission autosomique récessive. C’est-à-dire qu’elle se développe lorsque les deux parents transmettent les gènes défectueux à l’origine de cette maladie à leurs enfants. Il faut donc que les deux parents soient porteurs de cette variations du gène et qu’ils transmettent leur gène affectée. Les individus qui héritent d’une seule copie du gène muté sont des porteurs sains (comme leurs parents), sans symptômes de la maladie.
Le gène responsable de la mutation à l’origine de la maladie est localisé sur le chromosome 5 (5q13).
La prévalence de la maladie de Tay Sachs en Europe est d’environ 1 cas pour 320 000 naissance.
La maladie de Tay-Sachs partage les signes cliniques avec la maladie de Sandhoff et sont différenciées depuis peu de temps.
En 1881, l’ophtalmologiste britannique Warren TAY décrit la présence d’une tache rouge cerise sur la rétine d’un jeune enfant présentant un retard physique et mental. A la même période, le neurologue américain Bernard SACHS rapporte l’altération du développement cérébral chez des malades ayant un tableau clinique similaire. Cette maladie fut appelée maladie de Tay-Sachs.
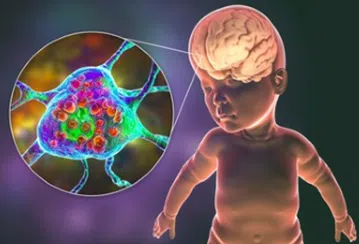
Dans les années 60, on découvre que la maladie est due à une accumulation d’une substance lipidique dans les cellules, la ganglioside GM2. Cette substance est normalement dégradée par une enzyme, appelée hexosaminidase A (HEXA), présente dans le lysosome. Cette enzyme HEXA est un assemblage de 2 sous-unités : une sous-unité alpha et une sous-unité béta. La mutation génétique du gène HEXA, génère un défaut dans la production de la sous-unité alpha qui va réduire l’activité de l’enzyme, limiter la dégradation de la ganglioside GM2 qui finit par s’accumler et surcharger les cellules du cerveau.
En fonction de la nature spécifique de la mutation HEXA, la quantité d’enzyme fonctionnelle produite peut varier, ce qui peut expliquer les différences dans la gravité des symptômes et l’âge d’apparition de la maladie. Les formes infantiles de la maladie de Tay-Sachs sont généralement associées à une absence presque complète d’activité enzymatique et sont les plus sévères, tandis que les formes juvéniles et adultes de la maladie sont généralement associées à une activité enzymatique résiduelle et ont des symptômes moins graves et une progression plus lente.
Les 3 formes de la maladie de Tay-Sachs
Pour la maladie de Tay-Sachs, il en existe trois formes cliniques selon l’âge de début :
– les formes infantiles précoces (type 1) débutent entre l’âge de 3 et 6 mois. Elle est la forme la plus fréquente et sévère. Il existe un déficit très important voire une absence d’activité de l’hexosaminidase A dans les leucocytes ou les fibroblastes cultivés après biopsie de peau.
– les formes juvéniles (type 2) débutent entre 2 et 10 ans. La diminution de l’activité de l’hexosaminidase A est moins importante que dans la forme infantile.
– les formes adultes (type 3) peuvent débuter à l’adolescence ou à l’âge adulte. Il existe un déficit en hexosaminidase A mais une activité résiduelle qui réduit la sévérité..
Deux variantes de la maladie ont été rapportées.
La gangliosidose à GM2 variante B1 présente un tableau clinique identique aux formes juvénile et adulte des variantes B « classiques ». Le déficit en hexosaminidase A ne peut se détecter qu’avec un substrat artificiel particulier, différent de celui de la variante B.
La gangliosidose à GM2 variante AB présente un tableau clinique d’une maladie de Tay-Sachs, mais l’activité de l’hexosaminidase A est normale. Il existe un déficit de l’activateur de l’enzyme nécessaire à l’hydrolyse du GM2.
La maladie de Tay-Sachs est un type de trouble du stockage lysosomal aussi appelé sphingolipidose. Elle est provoquée par une accumulation de graisses (lipides) dans le cerveau et la moelle épinière notamment.
Des sphingolipidoses se développent lorsque les personnes n’ont pas les enzymes nécessaires à la dégradation (métabolisme) des sphingolipides, qui sont des composés protégeant la surface cellulaire et remplissant certaines fonctions dans les cellules. Il existe de nombreux types de sphingolipidoses au-delà de la leucodystrophie métachromatique (maladies de Fabry, Gaucher, Krabbe, Niemann Pick A et B, leucodystrophie métachromatique, Sandhoff).















")