Quels sont les signes ?
La littérature médicale rapporte souvent une grande variation dans la chronologie et l’occurence des symptômes liés aux mutations sur les gènes CLN. Il est donc difficile de tirer des généralités sur les conséquences de ces maladies sur les conditions de chaque patient. Les informations présentées ici reflètent plutôt une observation moyenne, qui peut être plus ou moins éloignée de ce que les patients vivent au quotidien.
Voici quelques caractéristiques communes à toutes les formes de CLN :
Symptômes neurologiques : Les patients atteints de CLN présentent souvent une dégradation progressive de leurs fonctions motrices et cognitives, accompagnée de convulsions et, dans de nombreux cas, de la perte de la vision.
Progression de la maladie : Les CLN sont des maladies progressivement dégénératives, ce qui signifie que les symptômes s’aggravent généralement avec le temps. Dans de nombreux cas, la maladie progresse assez rapidement, conduisant à une incapacité grave et souvent à un décès prématuré.
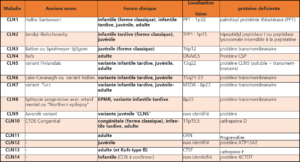
CLN1 – céroïdes lipofuscinoses neuronales 1
ou maladie de Santavuori-Haltia
L’âge d’apparition et la progression de la maladie peut varier énormément, mais on constate une corrélation entre les symptômes apparents, et la progression au sein de la cellule.
CLN1, forme infantile
- développement de la maladie en général entre 6 et 12 mois avec une hypotonie (faiblesse musculaire)
- l’enfant commence généralement à perdre ses compétences acquises vers 12 mois.
- perte de la parole, perd la compréhension de simples phrases.
- perte de la vision progressive, jusqu’à la cécité vers l’âge de 2 ans.
- détériorations du développement en général entre 15 et 20 mois.
- apparition des crises d’épilepsie en général vers 30 mois
- perte des capacités cognitives et motrices avant l’âge de 3 ans.
- la maladie conduit à un décès prématuré, généralement entre 9 et 13 ans.
CLN1, forme infantile tardif
- les premiers symptômes apparaissent enre 1,5 et 3,5 ans, avec un déclin de la cognition, une perte de la vision, de l’épilepsie.
- la progression de la maladie ressemble à celle de la CLN2.
- la maladie conduit à un décès prématuré, généralement entre 10 et 13 ans.
CLN1, forme juvénile
- la progression de la maladie ressemble à la CLN3 juvénile. L’épilepsie se développe cependant un peu plus tard, et les troubles moteurs un peu plus tôt que dans une CLN3 classique.
CLN1, forme adulte
- Maladie très rare, seulement deux patientes connues dans la littérature.
CLN2 – céroïdes lipofuscinoses neuronales 2
ou maladie de Jansky-Bielschowsky
- apparition des premiers symptômes entre 2 et 4 ans
- les premiers symptômes sont une diminution des capacités moteurs (des gestes maladroits, un manque de motricité fine = ataxie), et une déterioration de la parole, qui arrive généralement simultanément, avec une rapide progression vers 3 ans.
- les enfants sont généralement complètement dépendants à l’âge de 5 ans
- les crises d’épilepsie apparaissent généralement à la fin des trois ans. Leur forme peut varier suivant les patients. Il faut distinguer ces crises de la myclonie (contraction rapide des muscles), qui peut aussi apparaître, mais n’est pas traitée pareil. Ces crises de myclonie est un problème majeur qui empêche les enfants de dormir et se reposer.
- en général, à partir de 4 ans, la vision des enfants diminue progressivement, jusqu’à une cécité totale au bout de 3 ans. Chez certains patients cependant, les troubles de la vision n’apparaissent pas avant 10 ans
- beaucoup d’enfants perdent la capacité à déglutir, et doivent être équipés d’une sonde gastrique
- la maladie conduit à un décès prématuré, généralement au milieu des années d’adolescence.
CLN3 – céroïdes lipofuscinoses neuronales 3
Forme juvénile – maladie de Spielmeyer-Vogt-Sjögren ou maladie de Batten
- les premiers symptômes sont insidieux et liés à des pertes de capacités de la vision, qui sont généralement constatés entre 4 et 7 ans. Cela mène progressivement à une cécité totale, généralement au bout de 2 à 4 ans, mais la perception de la lumière peut être préservée pendant des années
- après la perte de la vue, les difficultés d’apprentissage à l’école deviennent évidents dès les premières années du primaire
- l’épilepsie se développe généralement à partir de 10 ans
- les symptômes moteurs arrivent généralement avec la puberté, avec d’abord des troubles de mouvement involontaires (syndrome extrapyramidal), et dans un moindre degré, des troubles des mouvements volontaires (syndrome pyramidal)
- la progression des symptômes moteurs amène progressivement à la perte de la mobilité indépendante
- des difficultés de la parole accompagnent généralement ces troubles moteurs, car la dysarthrie est un symptôme des troubles de mouvement involontaires
- des problèmes psychiatriques et de sommeil peuvent être observés à toute les périodes de la maladie
- la maladie conduit à un décès prématuré, en moyenne à l’âge de 24 ans (avec un intervalle constaté entre 10 et 28 ans) pour les patients atteints de la mutation 1kb.
- Une partie des documents disponibles sur la page du compte-rendu de la BDFA Family Conférence de 2016 concerne des informations spécifiques au quotidien des pré-adolescents atteints de CLN3.
- On pourra aussi consulter le rapport d’une étude menée sur 3 ans auprès de nombreux patients atteints de JNCL (variante juvénile de la CLN3). Le projet Erasmus+ s’intitulait Juvenile Neuronal Ceroid Lipofuscinosis and Education Project (2014 – 2017), et le rapport contient un nombre important d’informations sur les symptômes de la maladie. Pour aller plus loin, on peut
CLN4 – céroïdes lipofuscinoses neuronales 4
Forme de l’adulte ou maladie de Kufs
Le début a lieu vers 30 ans. Deux tableaux cliniques distincts ont été décrits :
- un phénotype A avec épilepsie myoclonique progressive, démence, ataxie, syndrome pyramidal et extrapyramidal, et
- un phénotype B avec troubles du comportement et démence au premier plan, puis apparition de signes cérébelleux et/ou extrapyramidaux.
L’évolution se fait dans les deux cas vers un état grabataire et le décès survient en moyenne dans les 10 ans après le début des signes.
CLN5 – céroïdes lipofuscinoses neuronales 5
Forme infantile tardive liée dit variant finlandais & forme juvénile précoce dite maladie de Lake et Cavanagh
Elle s’observe presque exclusivement en Finlande.
- apparition dés 4-5 ans par des difficultés de concentration, avec troubles des apprentissages scolaires, ou dans certaines variantes (en Colombie par exemple): 9 ans
- entre l’âge de 9 et 11 ans, progression rapide
- à partir de 12 ans, les enfants ont besoins d’aide pour leurs activités quotidiennes
- la maladie conduit à un décès prématuré entre 12 et 23 ans
- un trouble de la vision maculaire apparaît en premier, puis quelques années plus tard la cécité fonctionnelle est observée
- les premières crises d’épilepsie apparaissent en moyenne à 9 ans
- les capacités cognitives dès l’apparition des premiers symptômes visibles, et déclinent ensuite rapidement
- de légères difficultés motrices apparaissent dès l’âge de 3 à 6 ans, et les problèmes moteurs majeurs apparaissent entre 6 et 8 ans
- la plupart du temps, ces troubles ne sont pas accompagnés de troubles du comportement
- les troubles de la nutrition apparaissent généralement entre 9 et 13 ans
- la puberté apparaît généralement à un âge classique
CLN6 – céroïdes lipofuscinoses neuronales 6
Forme infantile tardive appelée variant turc
Il s’agit d’une variante rare de CLN infantile. Les symptômes cliniques sont en général très semblables à ceux observés pour une CLN2 / CLN5.
Cette forme est plus fréquente dans certains groupes, en particulier dans les populations indopakistanaises et probablement en Europe du sud (Portugal). Sont à noter :
- une régression psychomotrice à partir de 4 ans,
- des convulsions vers 5 ans,
- des myoclonies vers 5-6 ans (Contraction musculaire involontaire de courte durée),
- un déficit visuel vers 5-7 ans,
- une ataxie à partir de 4 ans (trouble de la marche et de l’équilibre),
- une perte de la marche vers 7 ans.
CLN7 – céroïdes lipofuscinoses neuronales 7
Forme infantile tardive appelée variant turc
Cette forme a été rapportée chez des patients d’origine turque.
Le tableau clinique est proche de la forme infantile tardive classique CLN2, sont à noter :
- un début vers 3-4 ans marqué par une régression psychomotrice,
- une épilepsie vers 4 ans,
- un déficit visuel à partir de 5 ans,
- une ataxie vers 3 ans (trouble de la marche et de l’équilibre),
- une perte de la marche vers 5-6 ans.
CLN8 – céroïdes lipofuscinoses neuronales 8
Forme infantile tardive Epilepsie progressive avec retard mental liée ou « Northern epilepsy »
Il s’agit d’un tableau clinique très particulier au sein du groupe des céroïde-lipofuscinoses. Le début a lieu dans la petite enfance et l’évolution est prolongée jusqu’à l’âge adulte.
La plupart des patients rapportés sont originaires du nord-est de la Finlande. Les symptômes apparents sont très variables pour les CLN8, dépendant du type de mutation.
Certains ont des symptômes correspondant à un CLN infantile tardif (début 2-4 ans), partiellement semblable aux CLN2. Une autre variante observée voit les symptômes apparaître entre 5 et 10 ans.
Le début est marqué par l’apparition de crises fébriles ou non, généralisées tonicocloniques (epilepsie) dans l’enfance avec une recrudescence à la puberté et une diminution à l’âge adulte.
Au niveau cognitif, on note une régression lente. Des anomalies motrices et une atteinte visuelle apparaissent après la puberté.
CLN9 – céroïdes lipofuscinoses neuronales 9
Cette forme n’existe plus, car les patients décrits avec cette maladie se sont avérés plus tard avoir une CLN5
CLN10 – céroïdes lipofuscinoses neuronales 10
L’une des formes est congénitale, elle commence in utero, avec des crises d’épilepsie. Les survivants ne vivent que quelques jours.
Une autre forme est infantile tardive, très rare, avec une perte de la vision nocturne à l’âge de 4 ans, puis des troubles de la coordination fine, et apparition de cécité partielle. Enfin, perte de la cognition, de la parole, des capacités moteur. À la mi-adolescence, de sévères retards mentaux.
CLN11 – céroïdes lipofuscinoses neuronales 11
forme récemment identifiée de forme adulte
CLN12 – céroïdes lipofuscinoses neuronales 12
forme récemment identifiée de forme juvénile
CLN13 – céroïdes lipofuscinoses neuronales 13
forme récemment identifiée de forme adulte, proche de la CLN4 Kufs type B
CLN14 – céroïdes lipofuscinoses neuronales 14
Forme récemment identifiée de forme infantile mais des médecins soutiennent que la maladie causée par des mutations de ce gène n’a pas les caractéristiques déterminantes d’une CLN.
















")

